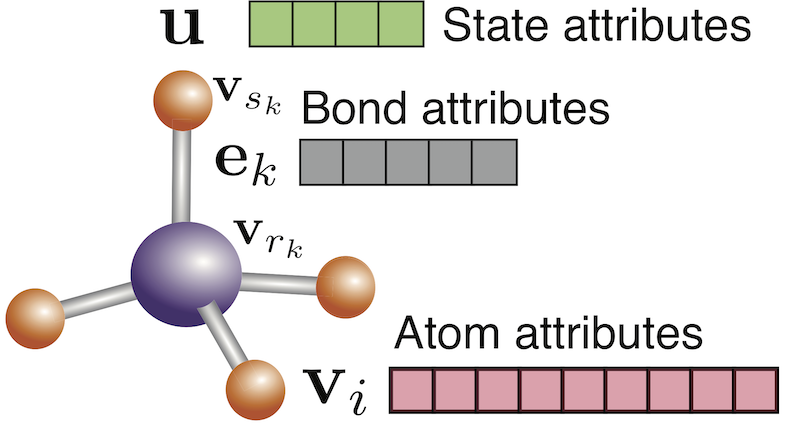
MRS Fall 2023 Tutorial on ML for SSBs
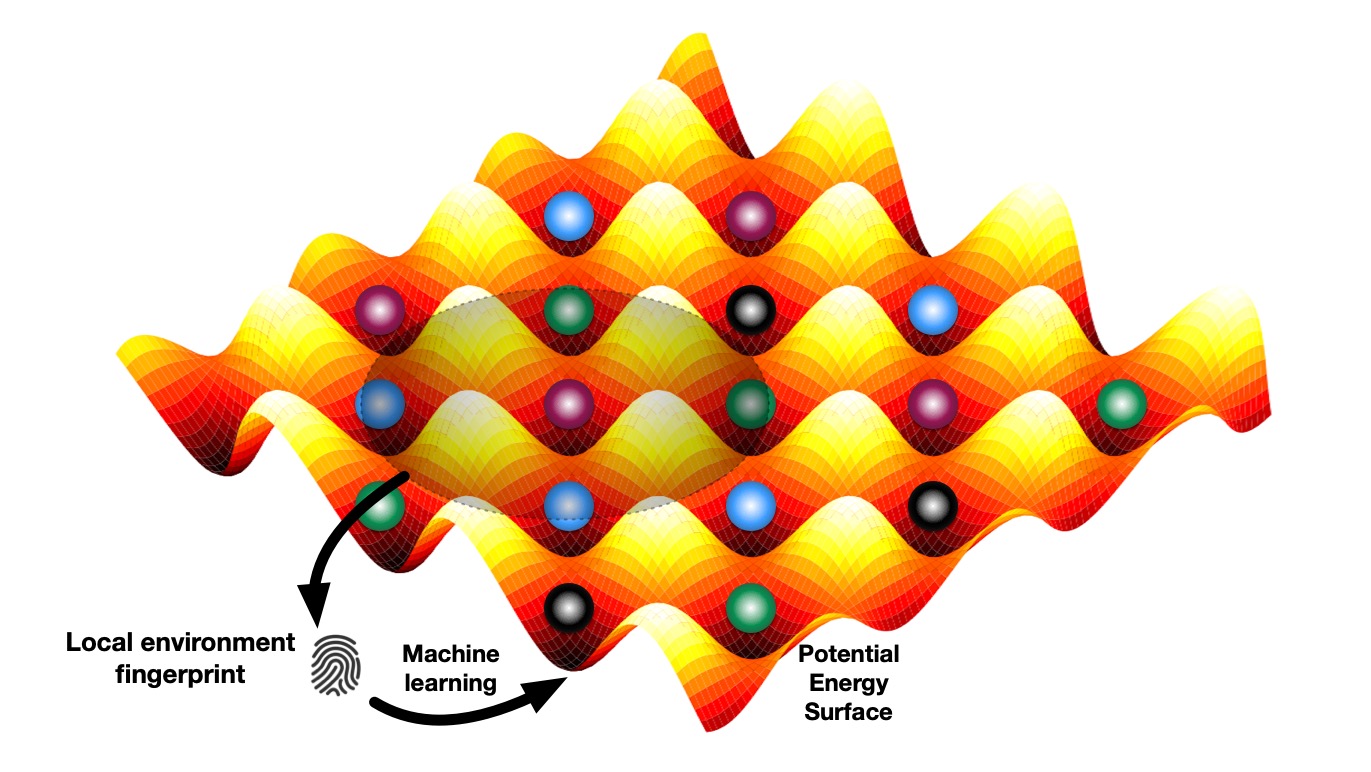
Ji Qi gave a tutorial talk on “Machine Learning and High-Throughput Discovery and Design of Next Generation Electrode and Superionic Materials and Their Interfaces for SSBs” at the MRS Fall 2023! This tutorial provides an overview of how our group is using ML techniques to gain insights and discovery alkali superionic conductors, as well as the many open-source software packages that we have developed for these purposes. A recording of this talk is available on our group’s YouTube channel (and embedded above).