CdS/V3O5 for Neuromorphic Computing
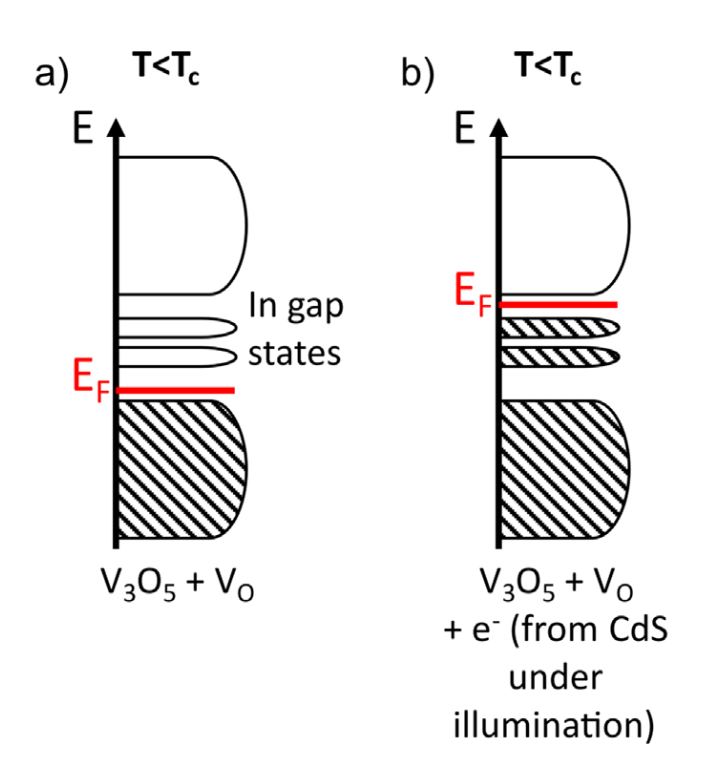
Congratulations to Jasleen on her first co-author paper on “An Optoelectronic Heterostructure for Neuromorphic Computing: CdS/V3O5” in Applied Physics Letters! Nonvolatile resistive switching is one of the key phenomena for emerging applications in optoelectronics and neuromorphic computing. However, the stochastic nature of the ion migration can be an impediment for the device robustness and controllability, with uncontrolled variations of high and low resistance states or threshold voltages. In this work, we report an optically induced resistive switching based on a CdS/V3O5 heterostructure. V3O5 is known to have a second order insulator to metal transition around 415 K, with an electrically induced threshold switching at room temperature. Upon illumination, the direct transfer of the photoinduced carriers from the CdS into V3O5 produces a nonvolatile resistive switching at room temperature. Jasleen’s contribution is in using DFT calculations to understand the defects present in V3O5 and the effects of electron doping. We show that electrons (generated by CdS under illumination) injected in V3O5 are trapped in a deep state, slowing the “low” temperature relaxation rate. For the LT phase (T < 340 K), the photoexcited electrons trapped into the oxygen vacancy are unable to overcome the barrier, and therefore, no relaxation is observed. […]