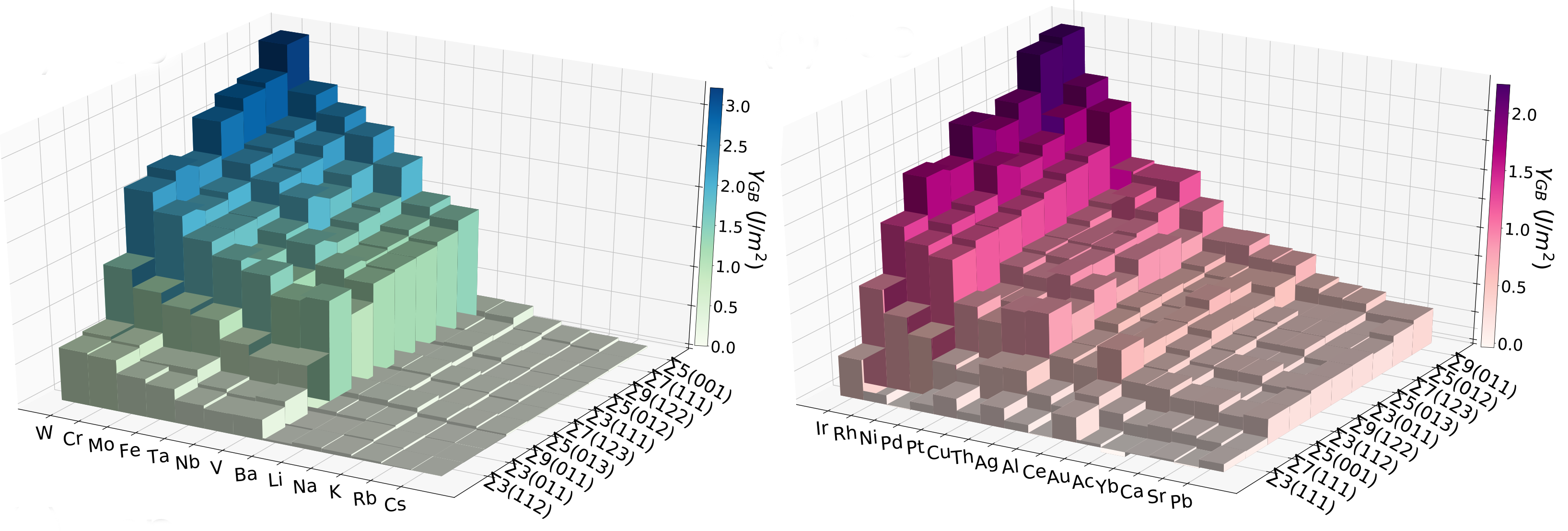
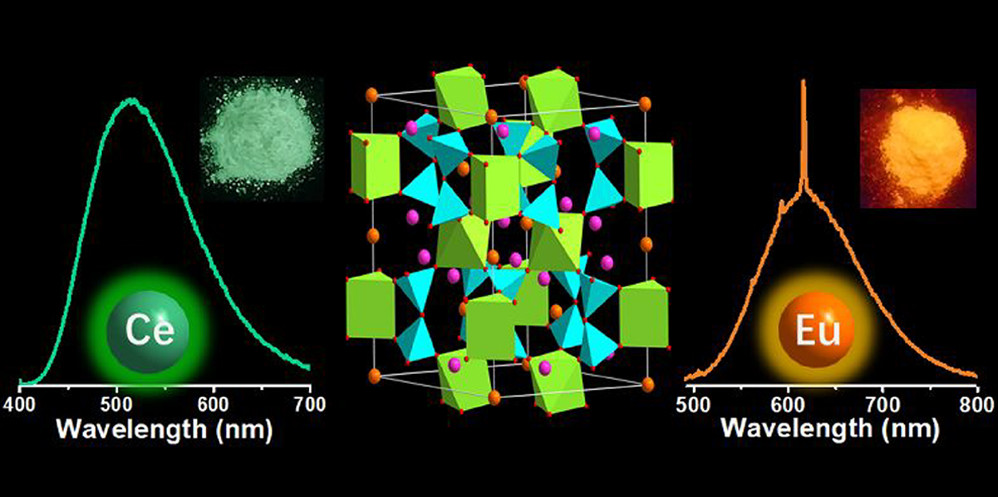
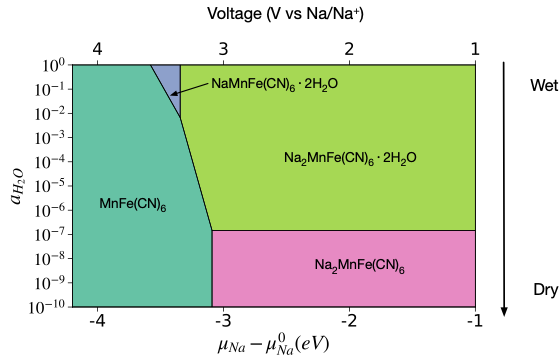
Random Forest Models for Coordination Environment from XANES

Chen Zheng’s and Chi Chen’s paper on “Random Forest Models for Accurate Identification of Coordination Environments from X-Ray Absorption Near-Edge Structure” has just been published in the second issue of the Cell Press journal Patterns! Analyzing coordination environments using X-ray absorption spectroscopy has broad applications in solid-state physics and material chemistry. Here, we show that random forest models trained on 190,000 K-edge XANES can directly identify the main atomic coordination environment with a high accuracy of 85.4%. More importantly, we show that the random forest models can be used to predict coordination environments from experimental K-edge XANES with minimal loss in accuracy. Check out the work here.