Electronic Structure 21 Workshop
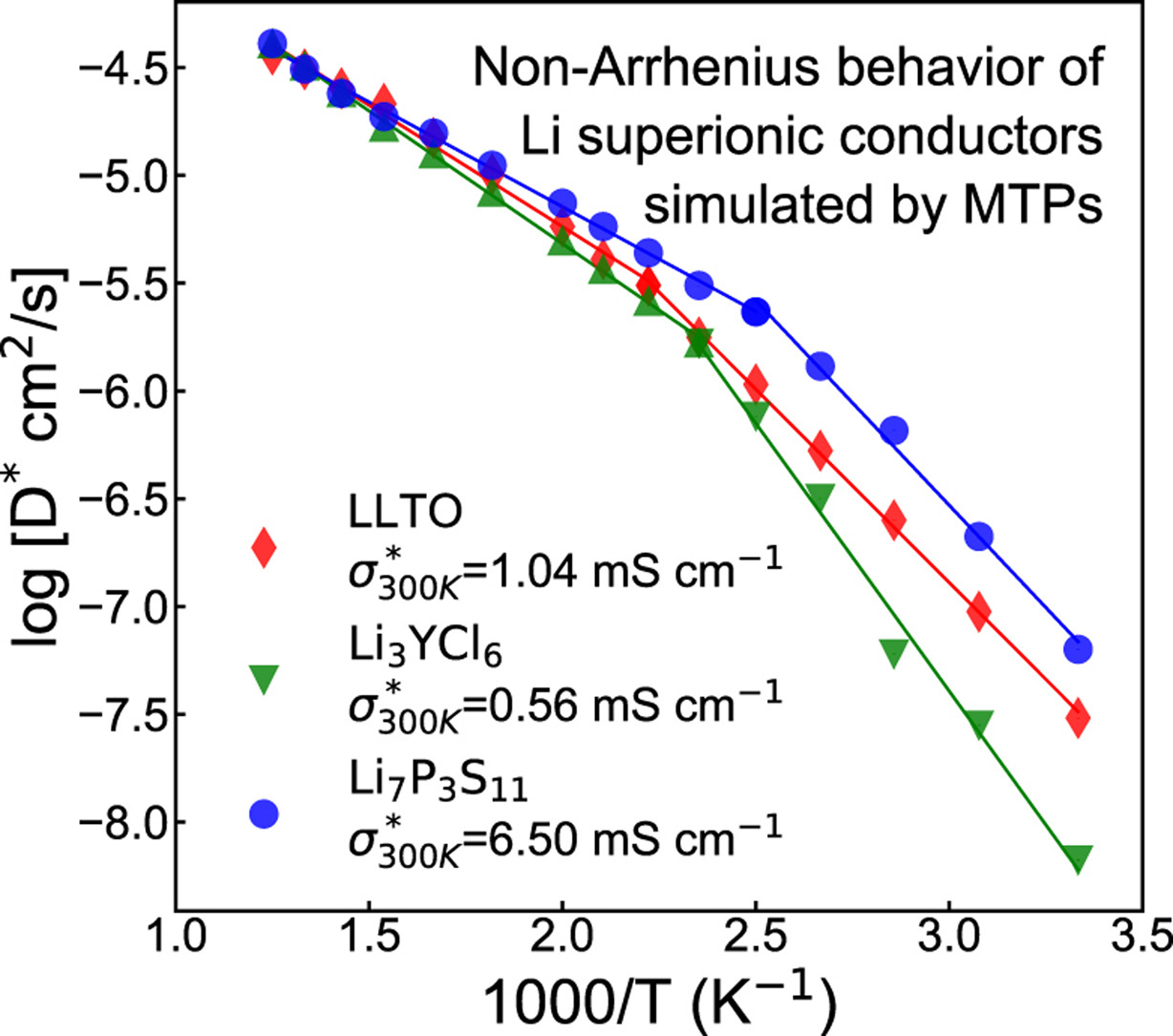
Prof Ong gave a talk on “Addressing Errors in Ab Initio Molecular Dynamics Predictions through Machine Learning” at the Electronic Structure 21 Workshop held on Jul 14 2021. In this talk, we discuss the errors that AIMD simulations make in predicting the properties of lithium superionic conductors, which can be traced to the small simulation cell sizes, high temperatures and short simulation times. We then demonstrate how machine learning can be used to construct highly accurate interatomic potentials (ML-IAPs) to enable long time scale, large system simulations of complex materials. In particular, a standardized approach to construct ML-IAPs for lithium superionic conductors such as Li7P3S11, Li3YCl6 and Li0.33La0.56TiO3 (LLTO) is shown. All three superionic conductor solid electrolytes exhibit a transition between Arrhenius regimes are relatively low temperatures due to a change in diffusion dimensionality.