Unlocking Superionic Sodium Transport and Synthesis with MLIPs
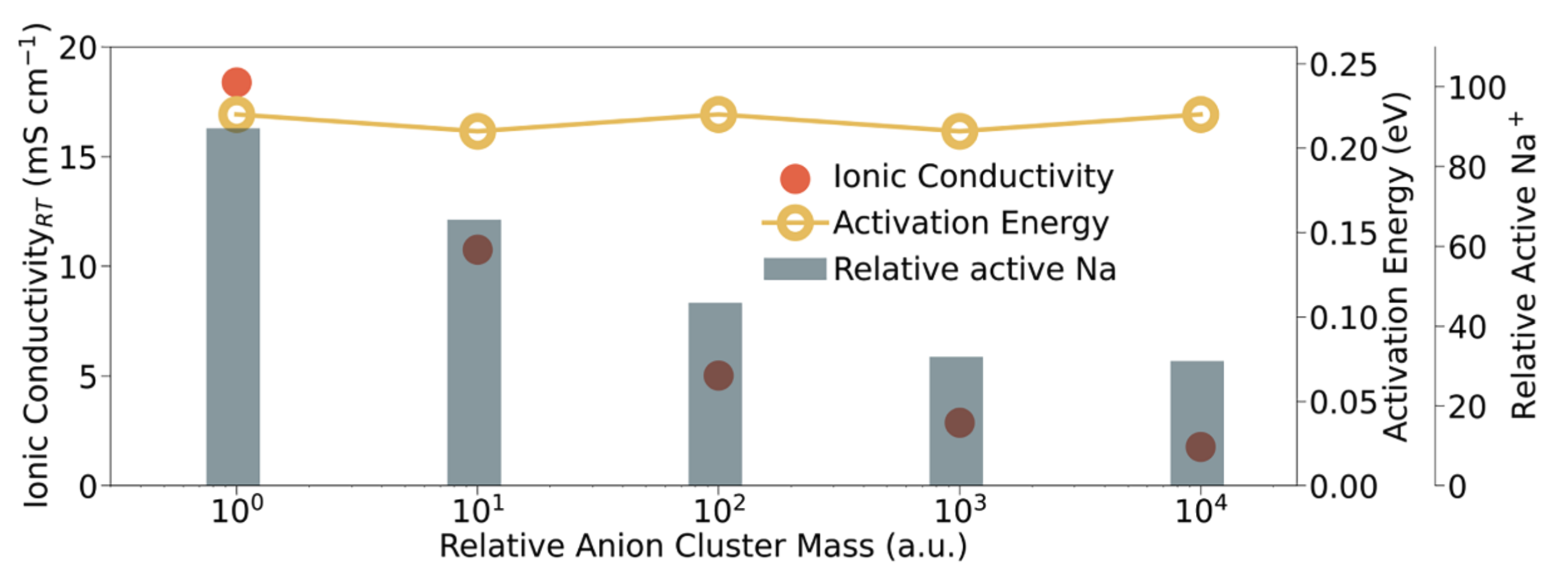
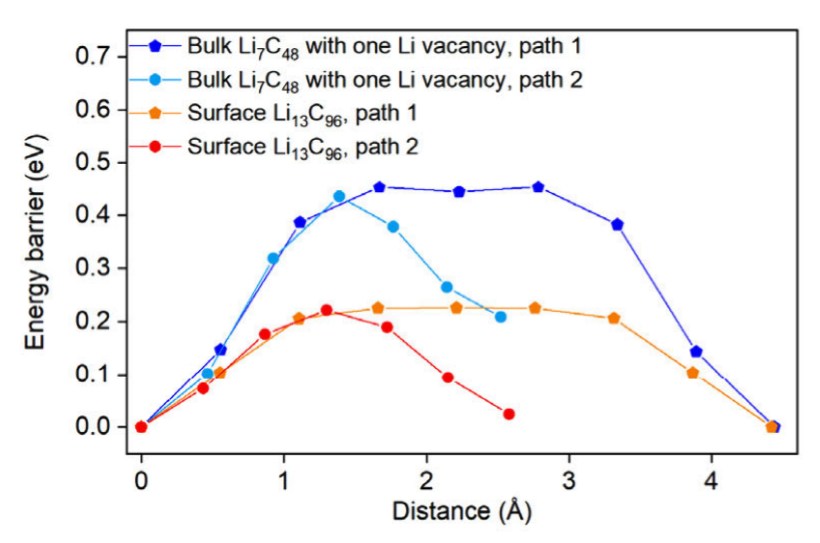
All-solid-state sodium batteries are attractive for grid-scale storage, but the search for a solid electrolyte that combines high ionic conductivity, mechanical compliance, and electrochemical stability has been challenging. In this collaboration with the Meng group published in Joule, we demonstrate that a metastable orthorhombic sodium closo-hydridoborate, Na₃(B₁₂H₁₂)(BH₄) (o-NBH) achieves 4.6 mS cm⁻¹ room-temperature conductivity and enables thick-cathode Na-ASSBs with >3 mAh cm⁻² areal capacity. Our group’s main contribution to this work is in the application of state-of-the-art machine learning interatomic potentials (MLIPs) in guiding synthesis and probing ion transport. MAVRL member, Zihan Yu, used using high-throughput r2SCAN density-functional theory (DFT) calculations to create a dataset and fine-tune a M3GNet foundation potential for the Na-B-H system. This MLIP allowed the generation of finite-temperature phase diagrams with phonon-derived vibrational entropies. These calculations revealed taht o-NBH sits ~16 meV atom⁻¹ above the 0 K hull but is entropically stabilized above ~650 K, in agreement with experiments. MD simulations with the fine-tuned M3GNet potential shows that o-NBH exhibits 3D Na⁺ diffusion pathways with activation energies of 0.13 eV (short-range) and 0.23 eV (long-range), consistent with NMR relaxation experiments. One major innovation is the use of artificial anion masses to probe the effect of anion rotation […]