Zr strengthening of MoSi2
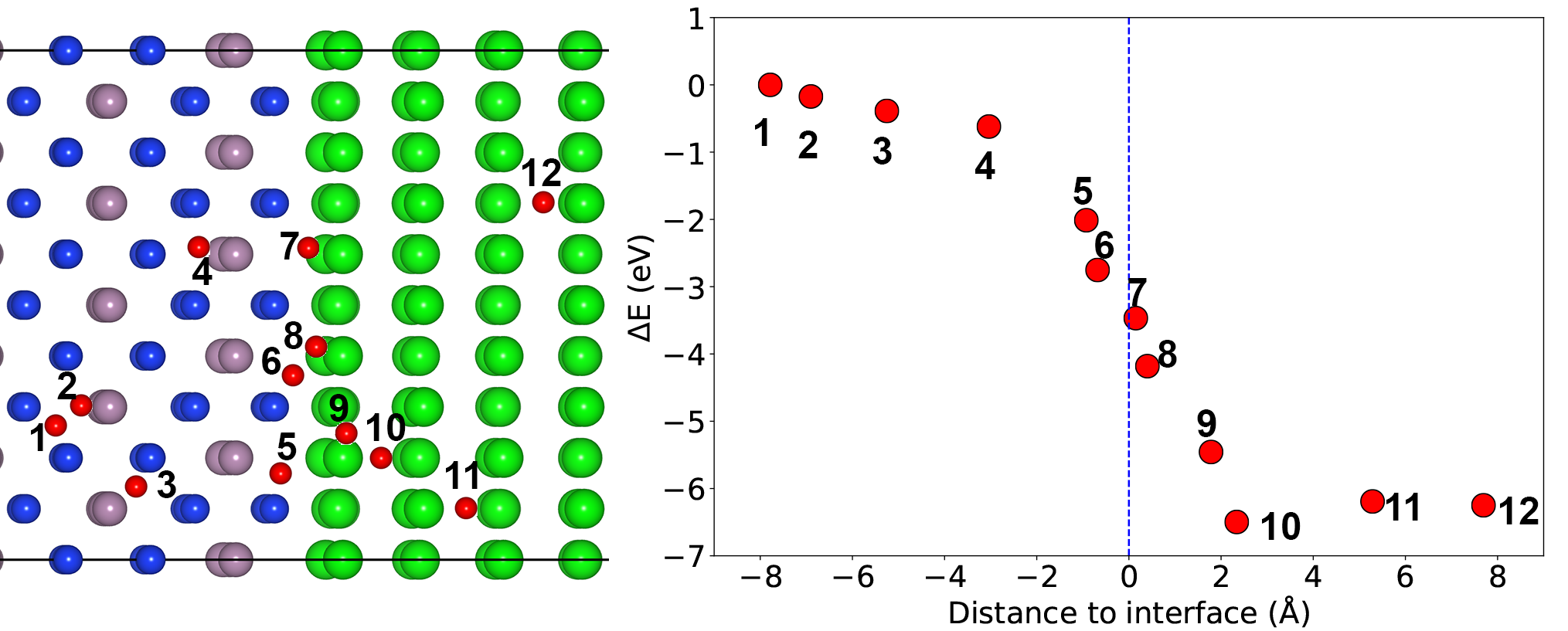
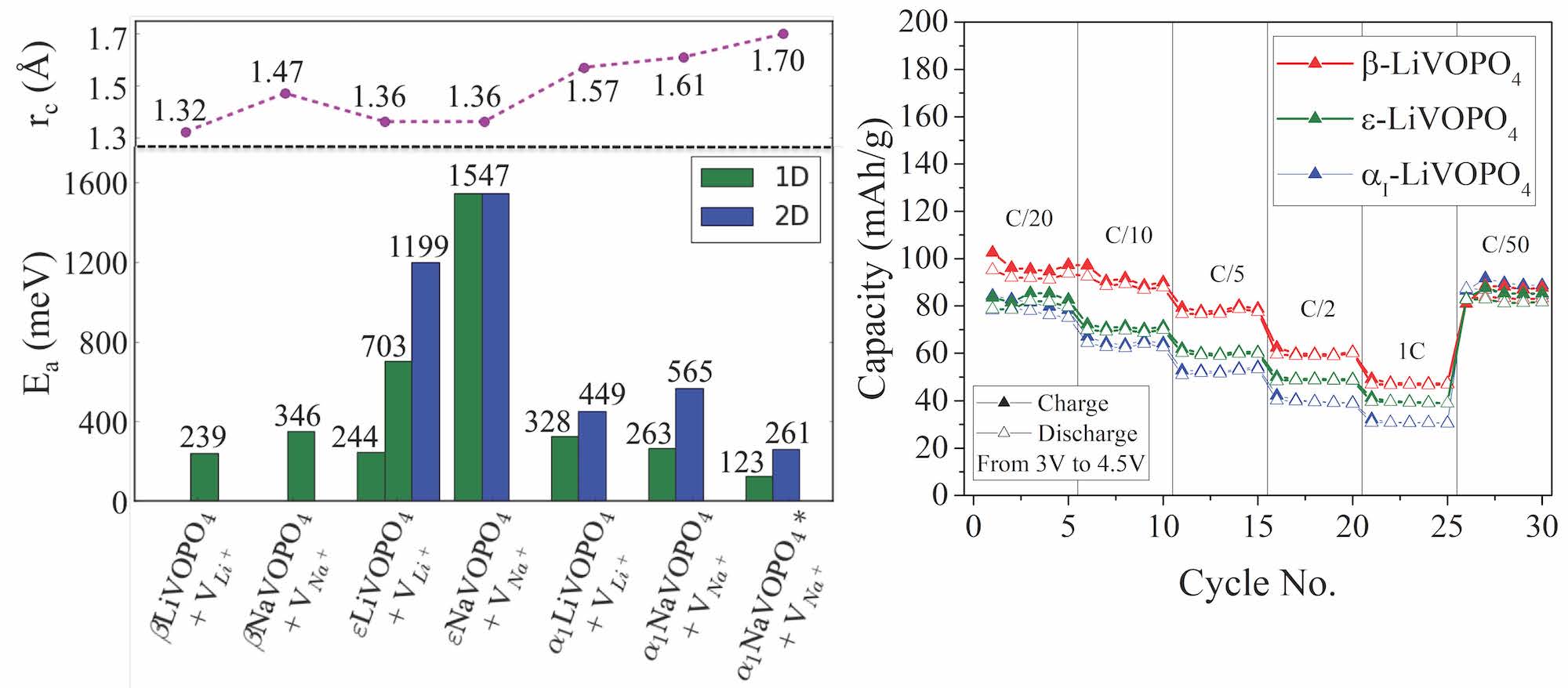
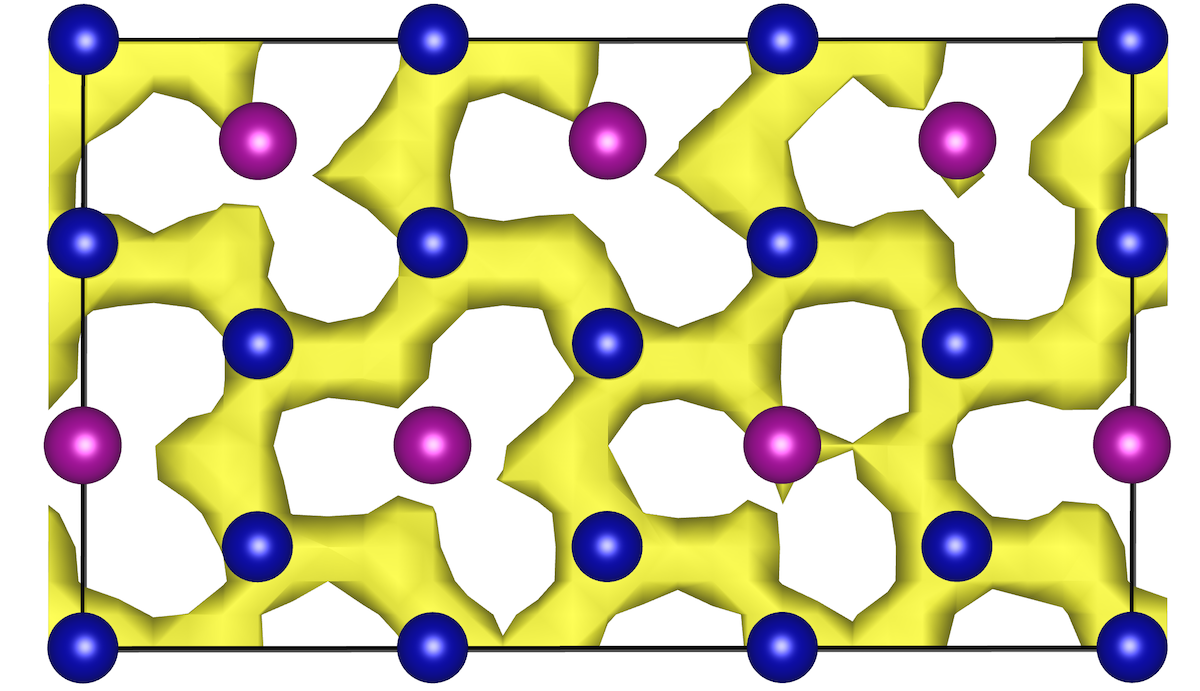
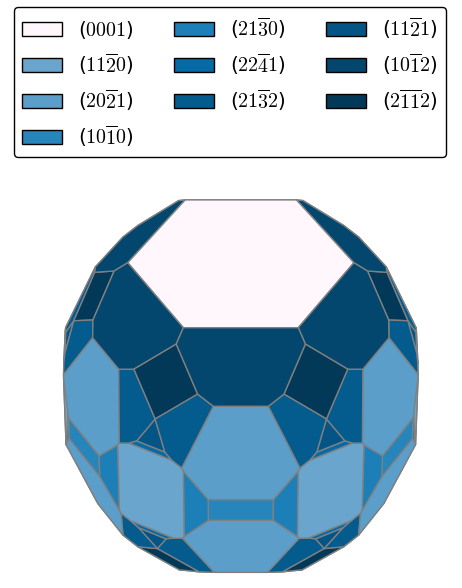
Hui Zheng’s first paper on “Role of Zr in strengthening MoSi2 from density functional theory calculations” has just been published in Acta Materialia. MoSi2 is an important intermetallic with excellent oxidation resistance at high temperatures. However, “pesting” by oxygen limits its application at intermediate temperatures. Using DFT calculations, we show that Zr nanoparticles act as a getter for oxygen, and in the process, significantly strengthens MoSi2 interfaces and grain boundaries. We also use the Materials Project to efficiently screen for other potential getter elements using simple thermodynamic descriptors, a general approach that can be extended to other alloy systems of interest.