
Chi’s Talk at the Global XAS Journal Club
Dr Chi Chen gave a talk at the Global XAS Journal Club on the Materials Virtual Lab’s efforts at constructing large X-ray absorption spectra databases using high-throughput computation and the development of machine learning models that can supercharge the interpretation of such spectra.
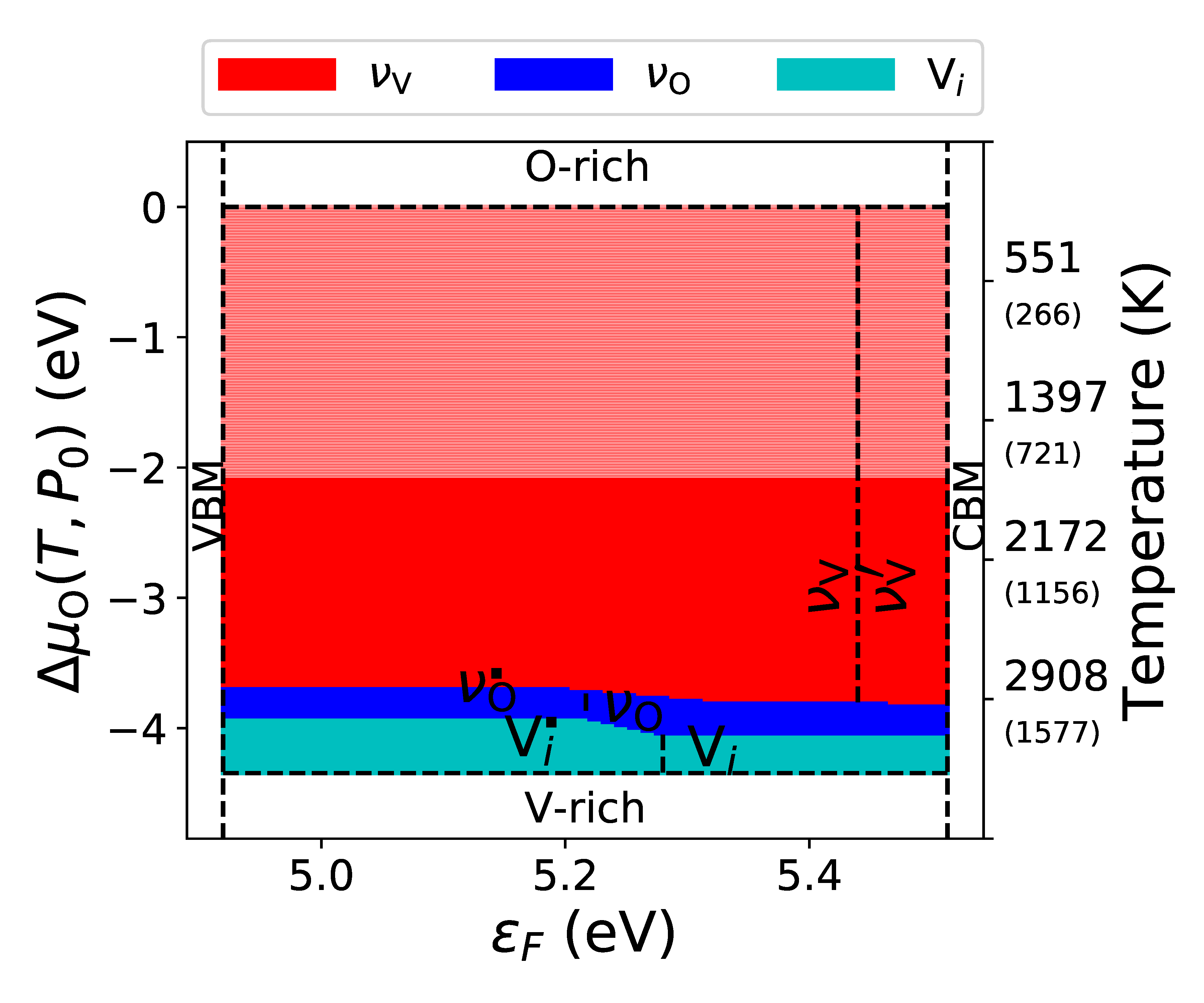
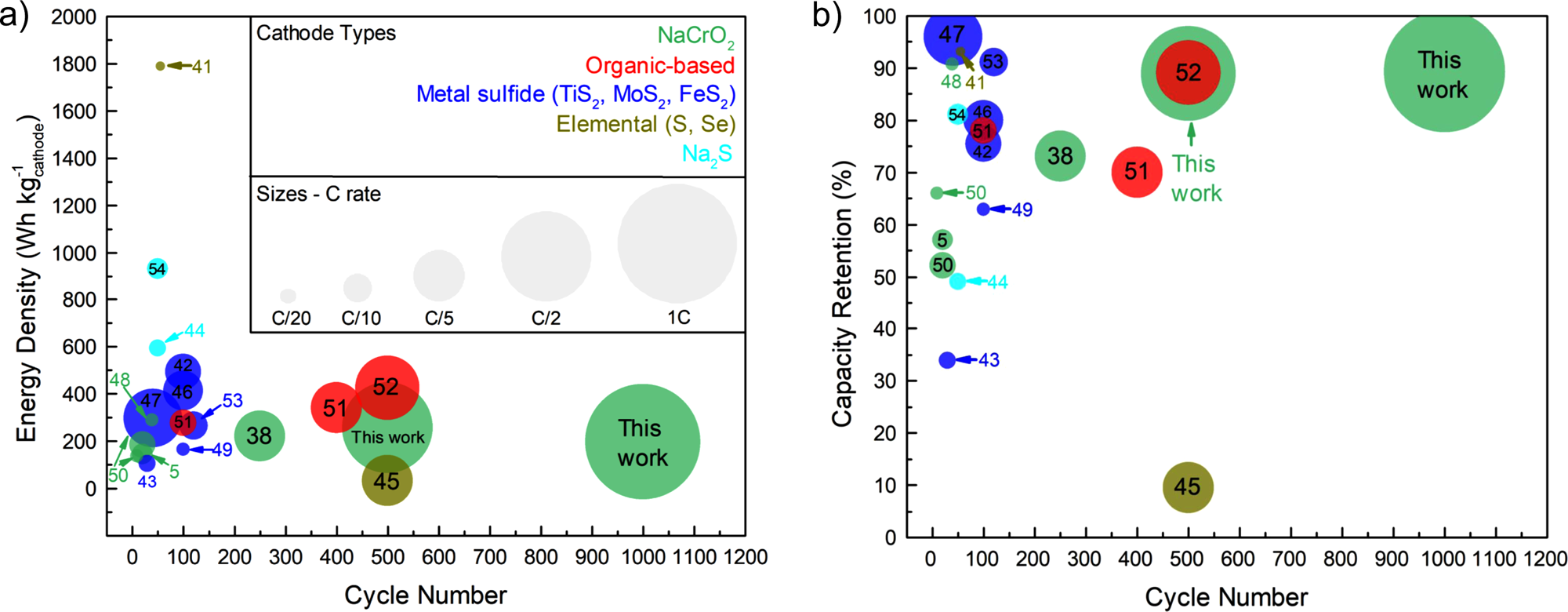
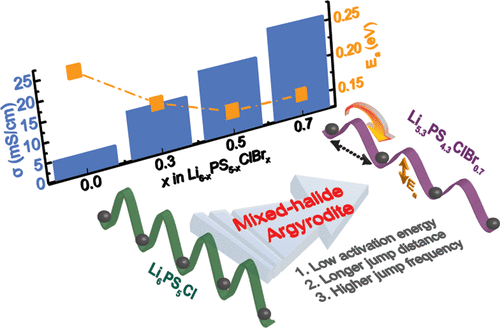
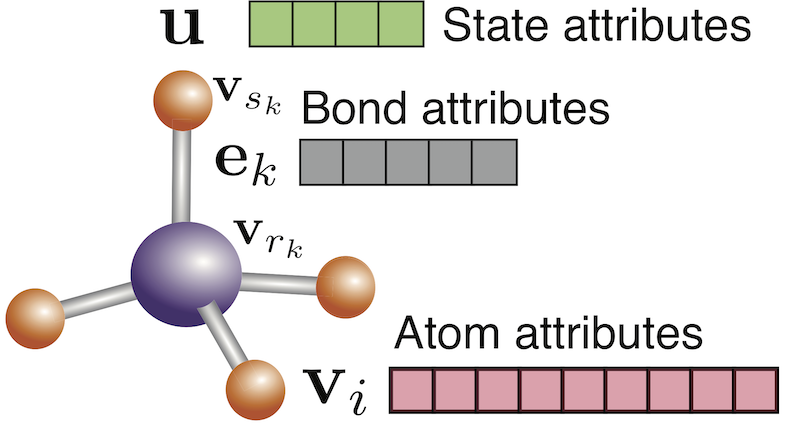
You must be logged in to post a comment.