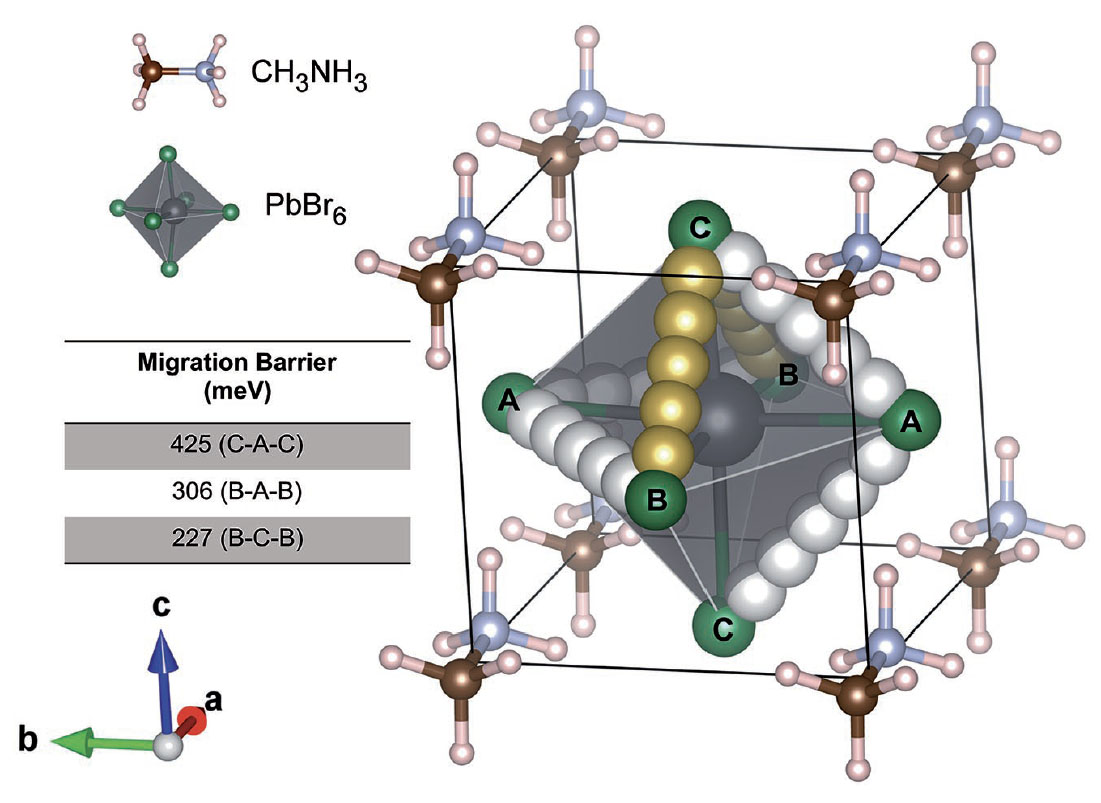
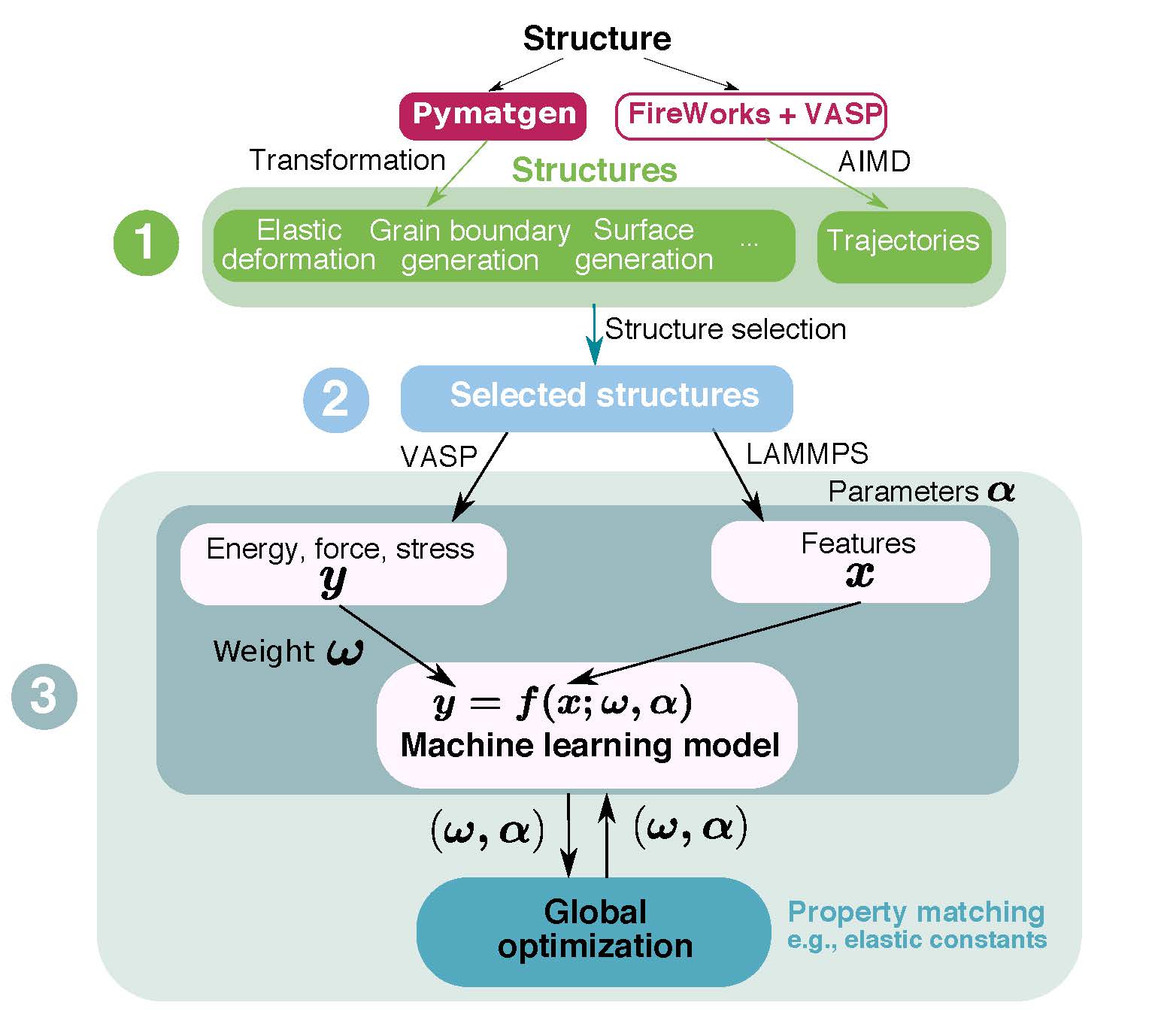
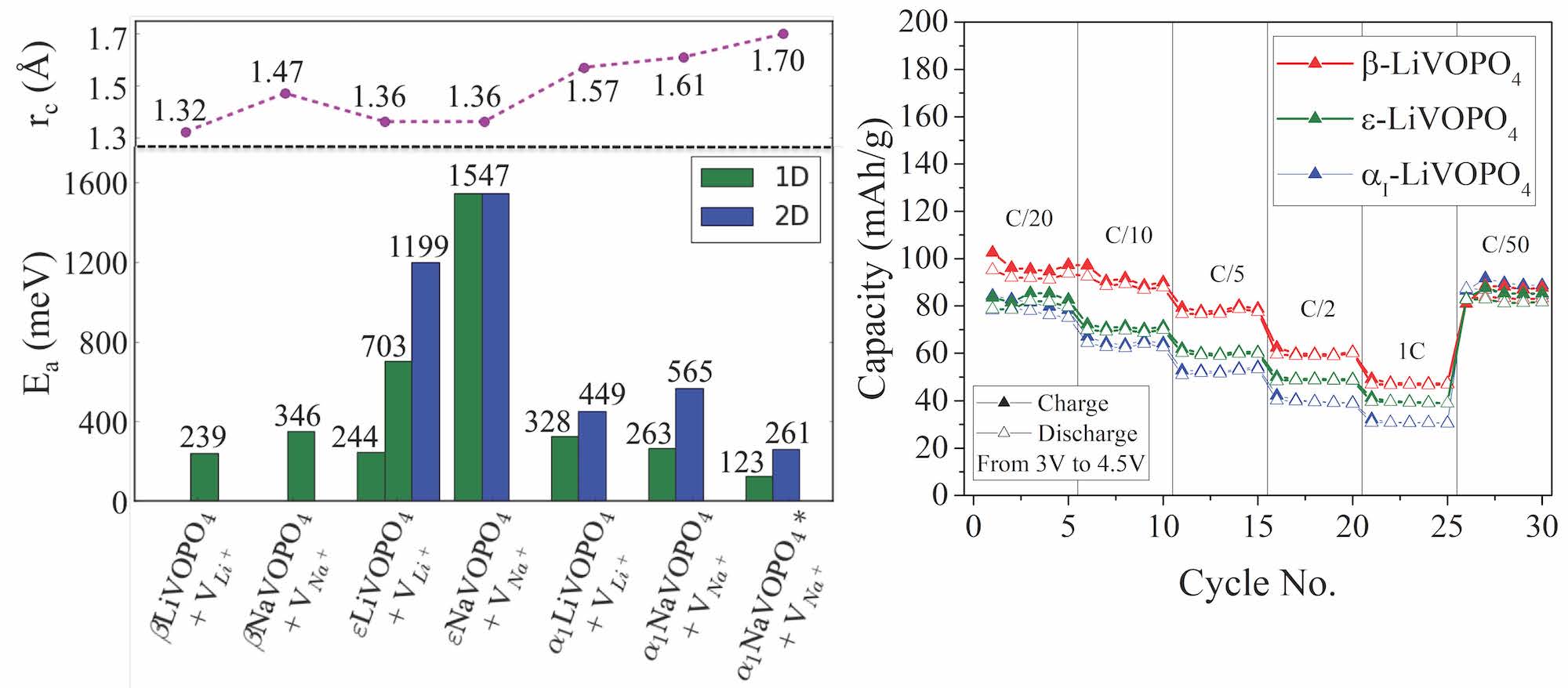
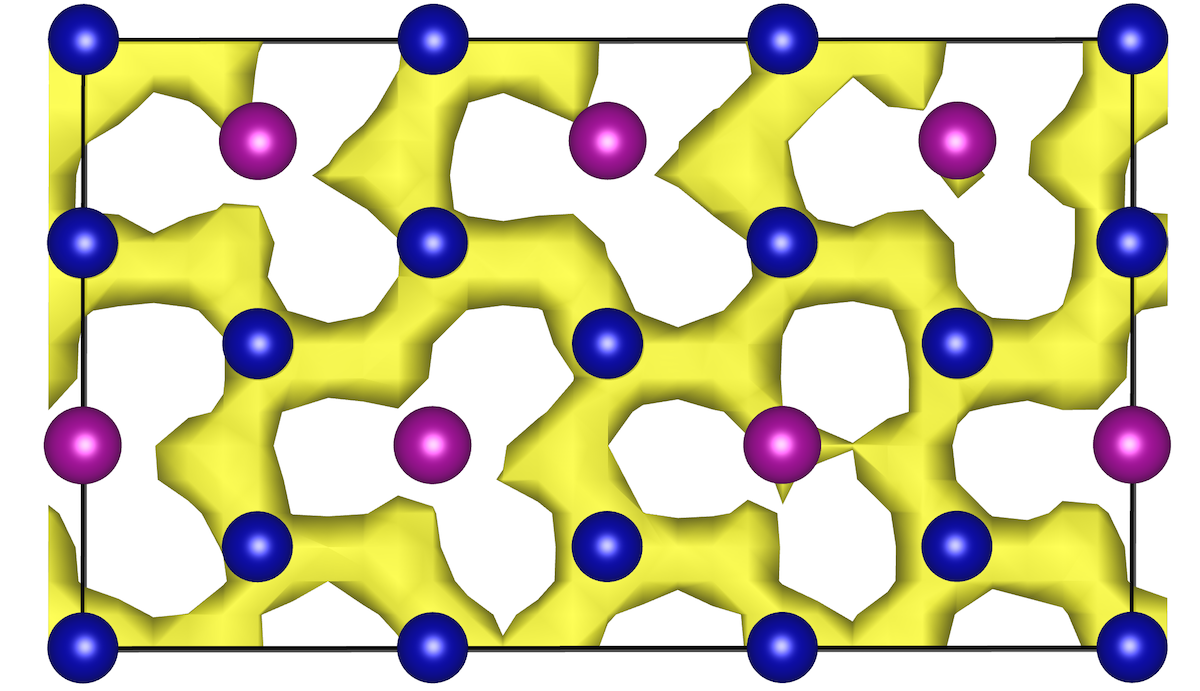
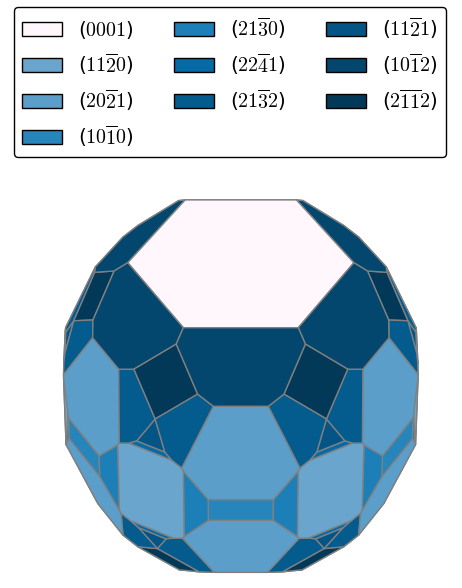
Book Chapter on Ab Initio Molecular Dynamics Studies of Fast Ion Conductors
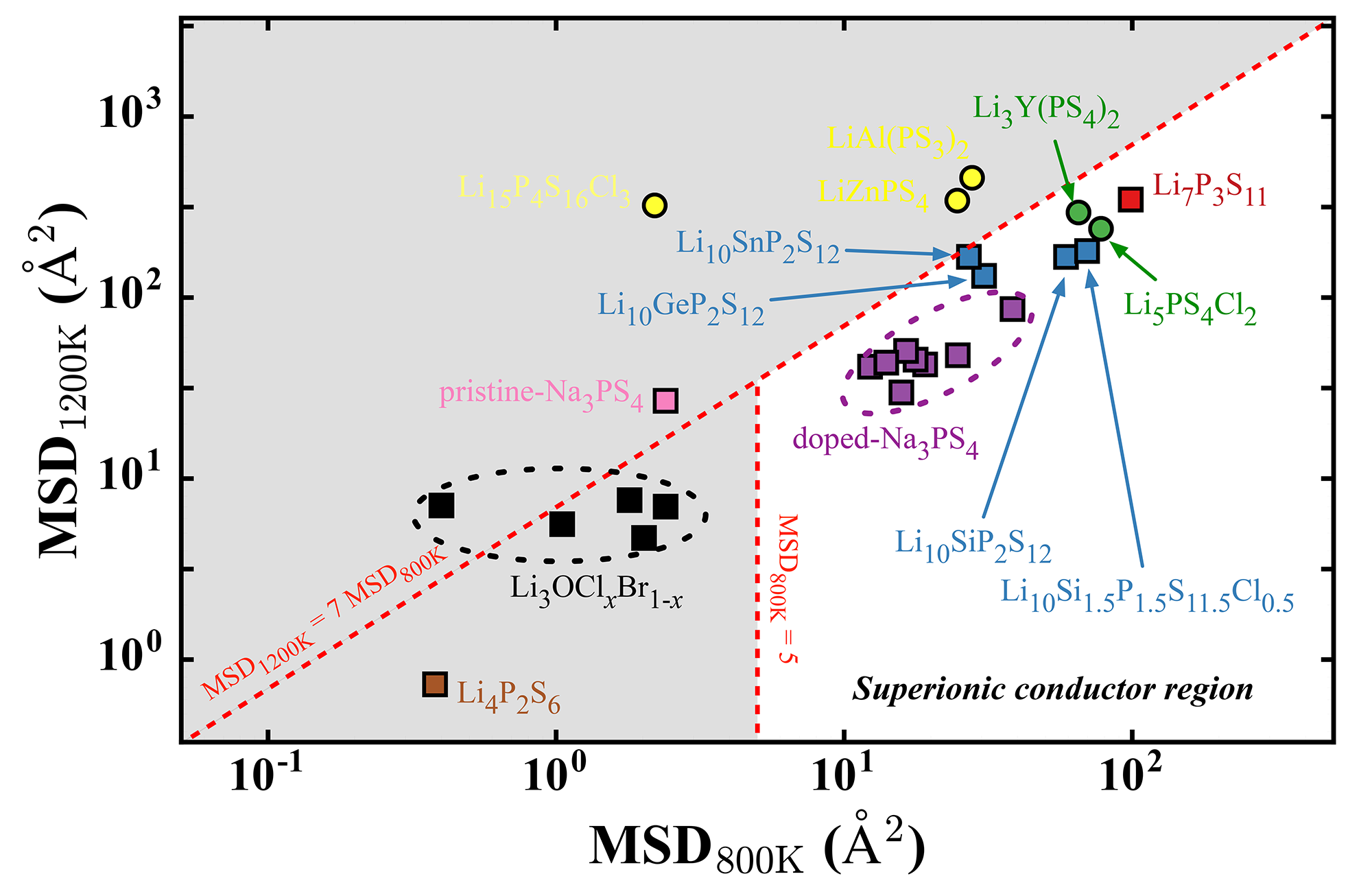
Our chapter on Ab Initio Molecular Dynamics Studies of Fast Ion Conductors in “Computational Materials System Design” is now available via Springer International Publishing.