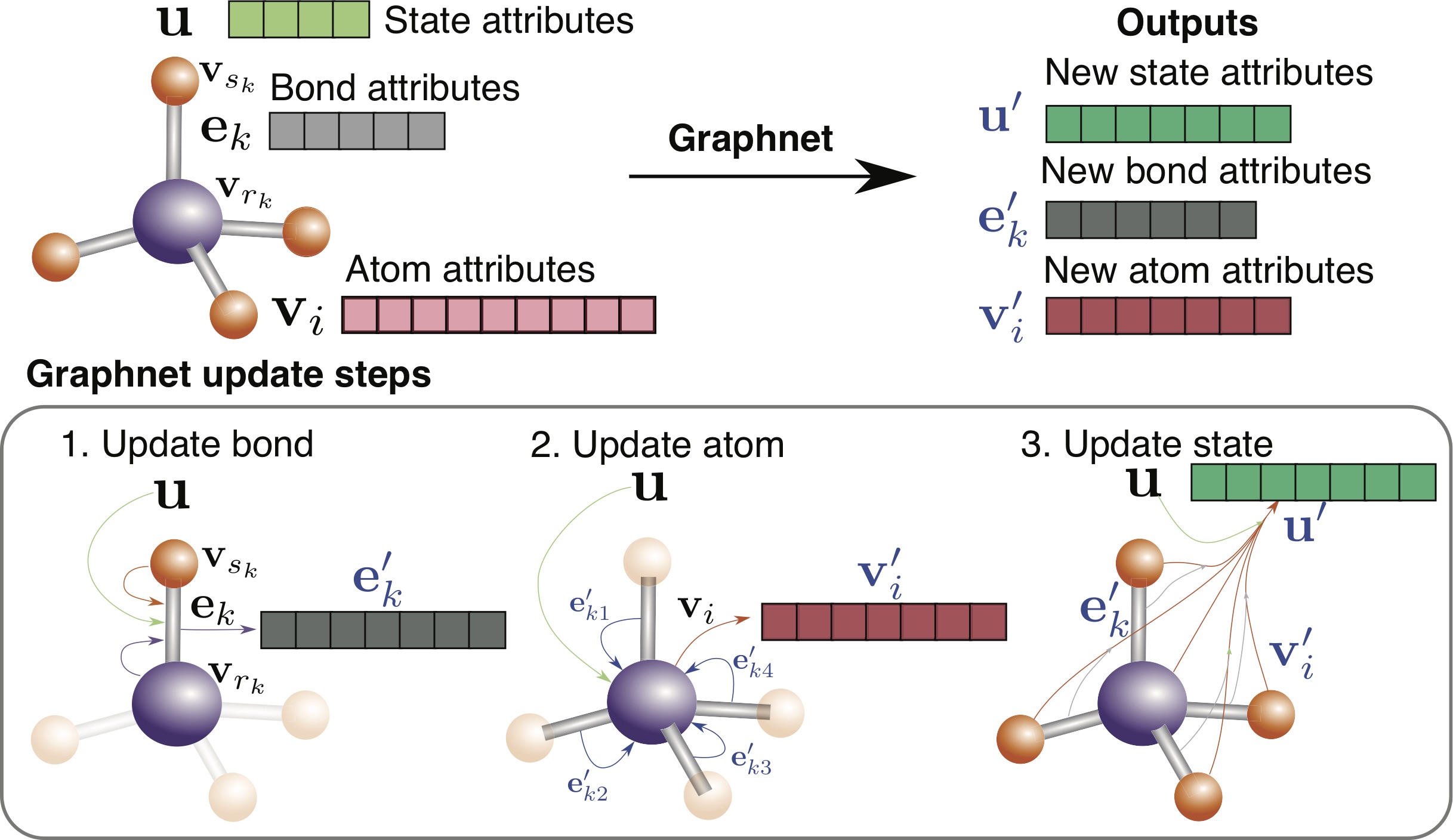
Database of Elemental Work Functions
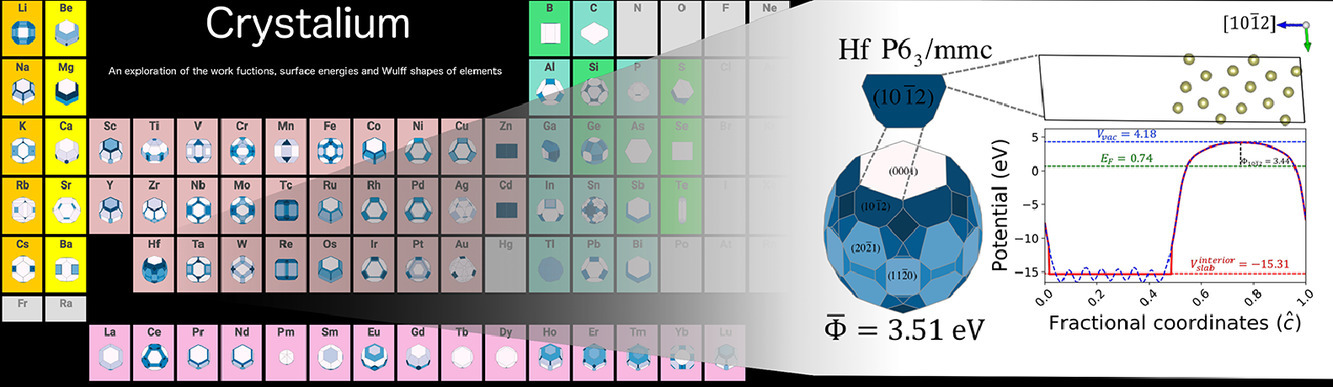
We are pleased to announce that Richard’s follow-up work on the anisotropic work functions of the elements has been published in Surface Science. The work function is a fundamental electronic property of a solid that varies with the facets of a crystalline surface. It is a crucial parameter in spectroscopy as well as materials design, especially for technologies such as thermionic electron guns and Schottky barriers. In this work, we present the largest database of calculated work functions for elemental crystals to date. This well-validated database contains the anisotropic work functions of more than 100 polymorphs of about 72 elements. One significant advance is the development of an improved model for the work function of metals from atomic parameters such as the electronegativity and metallic radius based on Gauss’ law.
The work function database can be accessed at the Crystalium website together with other surface properties.