
Machine learning properties of ordered and disordered materials
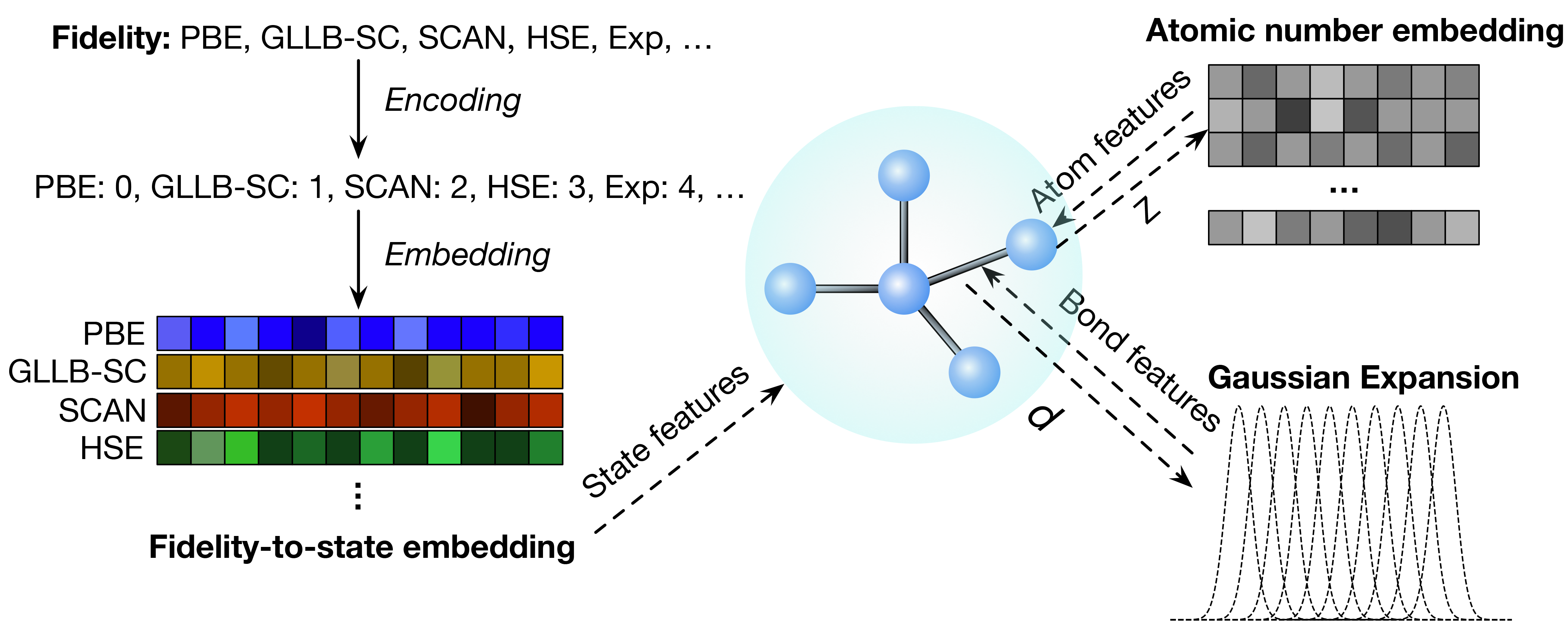
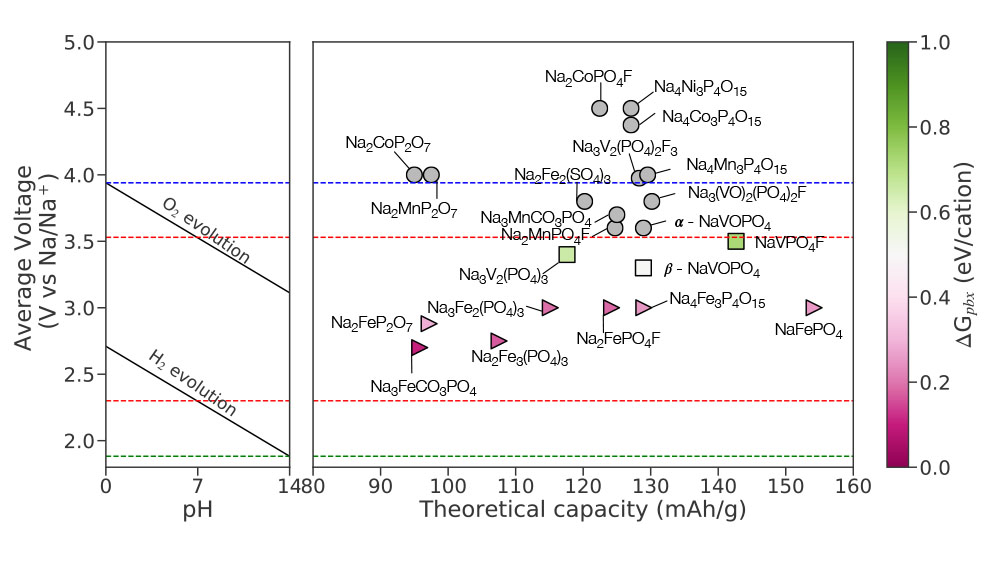
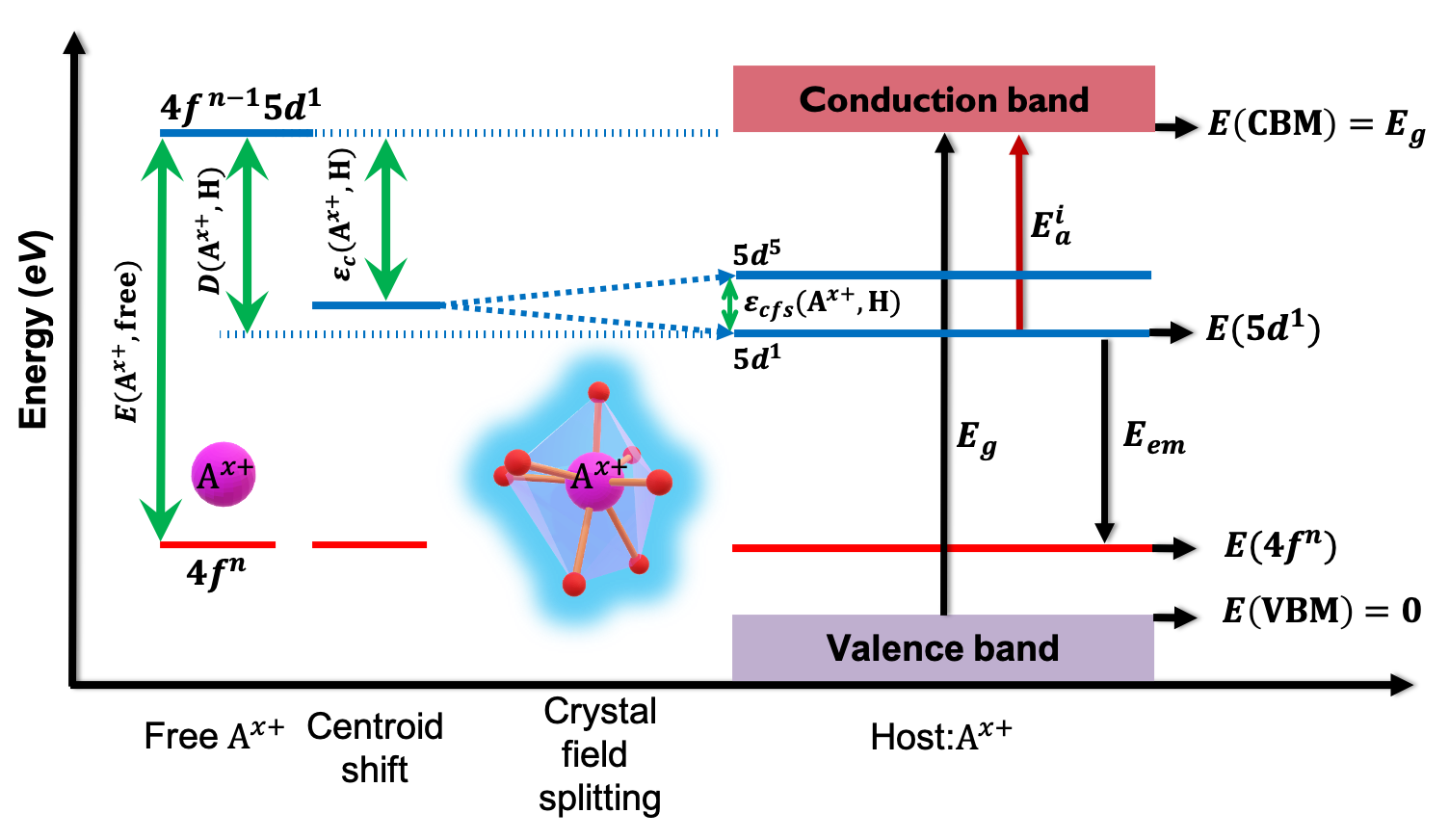
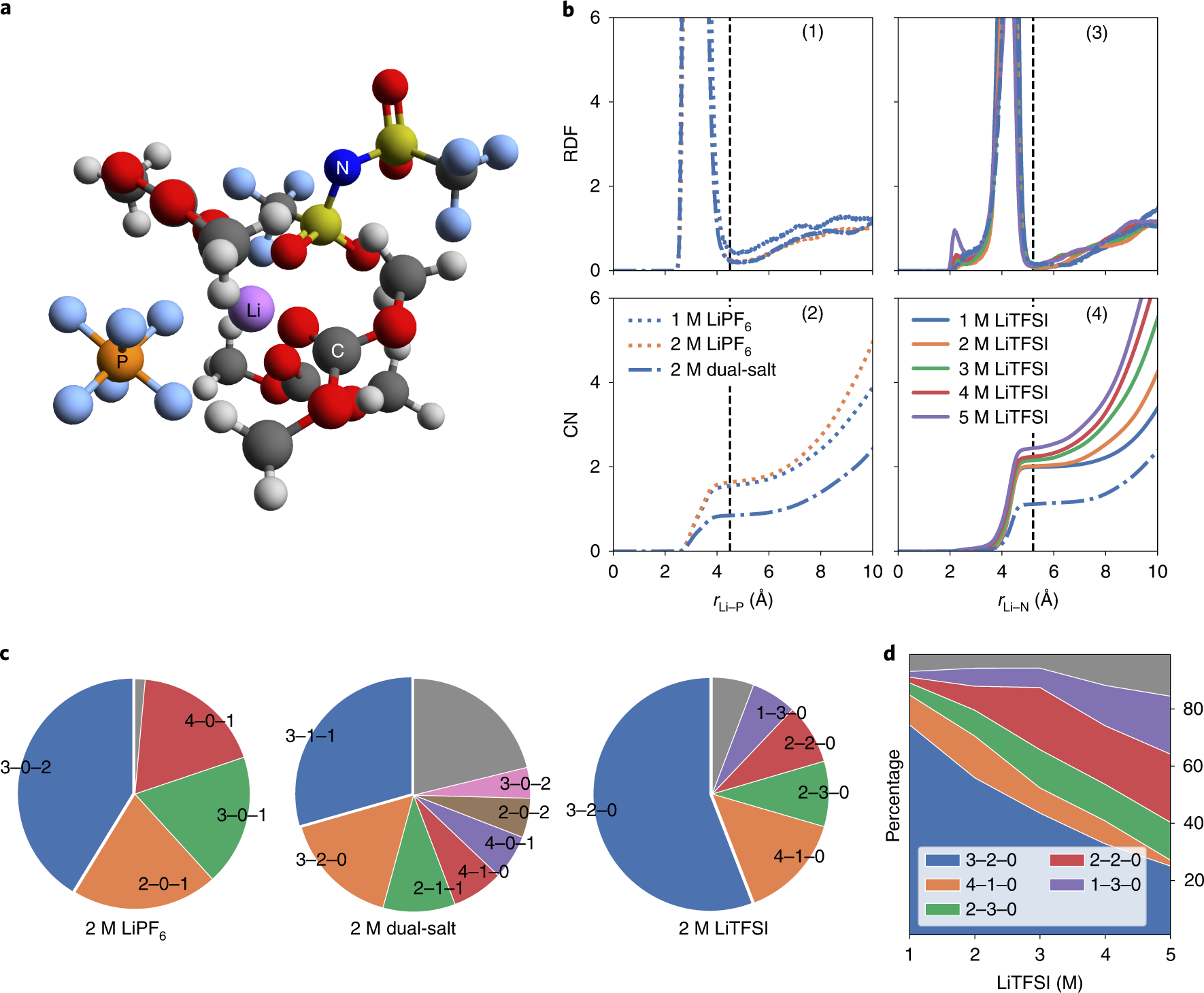
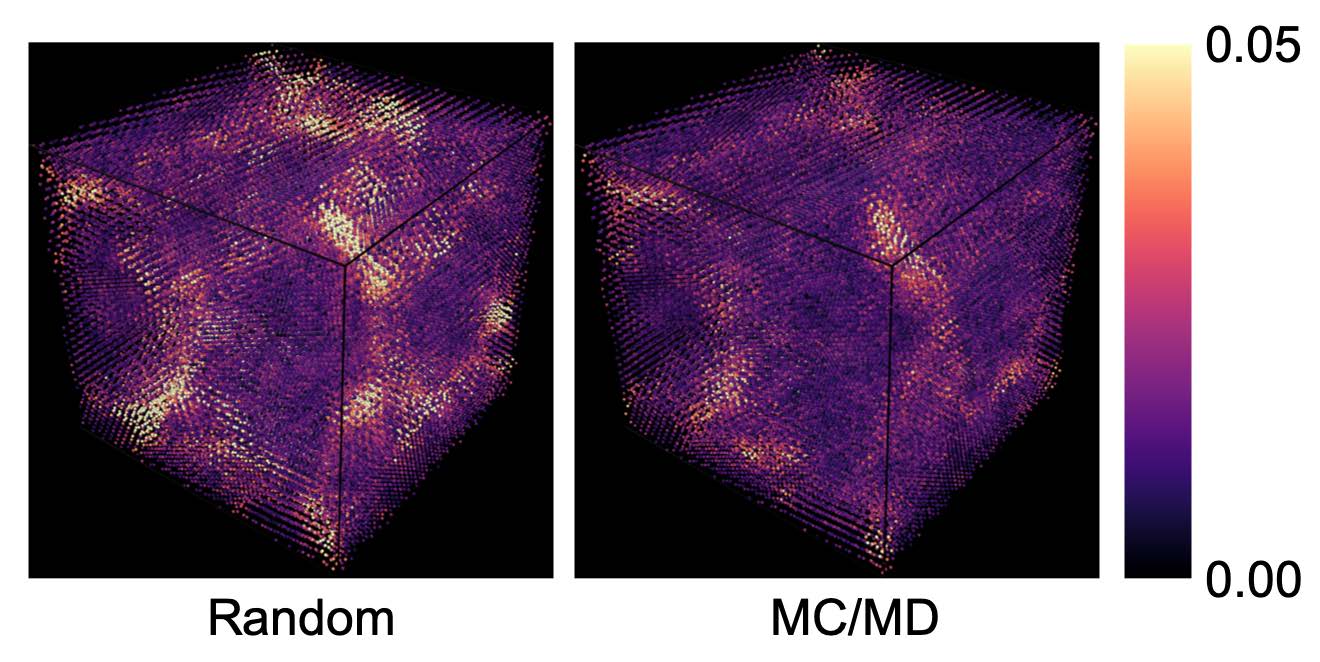
Our paper on “Learning properties of ordered and disordered materials from multi-fidelity data” has just been published in the inaugural issue of Nature Computational Science! In this work, we address two major impediments to ML for materials science. The first impediment is that valuable accurate data is much more expensive to obtain than less accurate data. Using multi-fidelity materials graph networks (MEGNet), we show that we can use the lower quality data to improve underlying structural representations in models, and in the process significantly improve predictions on smaller, more valuable data (e.g., experimental measurements). The second impediment is that making predictions on disordered materials, which is the vast majority of known materials, is much more difficult than on ordered materials. We show that the elemental representations (embeddings) learned by our MEGNet models can be used to directly model disordered materials. The article is available here. For an independent perspective on the findings, check out the Nature News & Views article. All data and code are available from http://crystals.ai and the Github repository.
You must be logged in to post a comment.