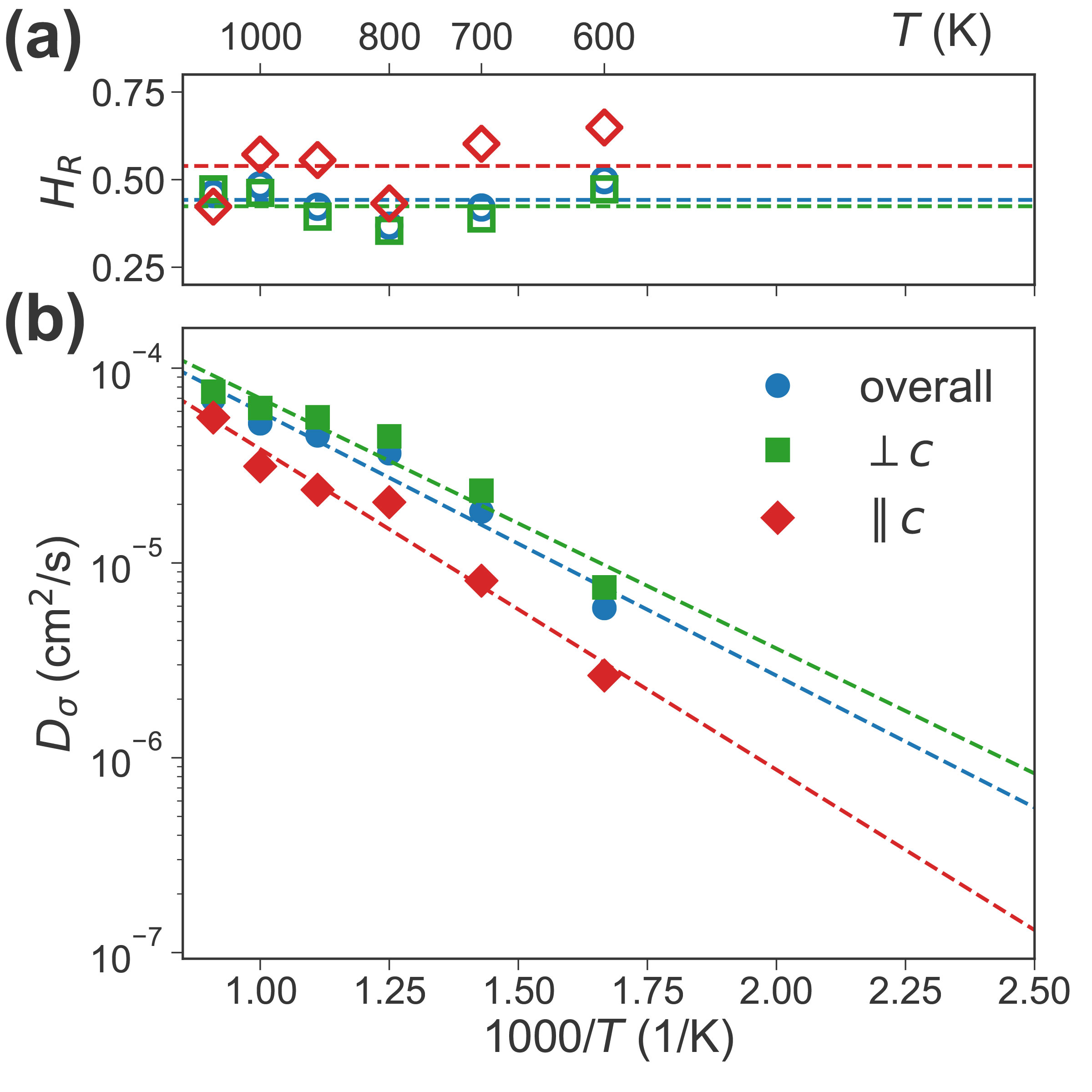
Water Contributes to Higher Energy Density and Cycling Stability of Prussian Blue Analogue Cathodes for Aqueous Sodium-Ion Batteries
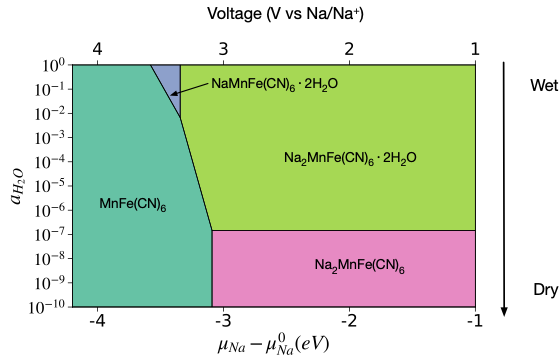
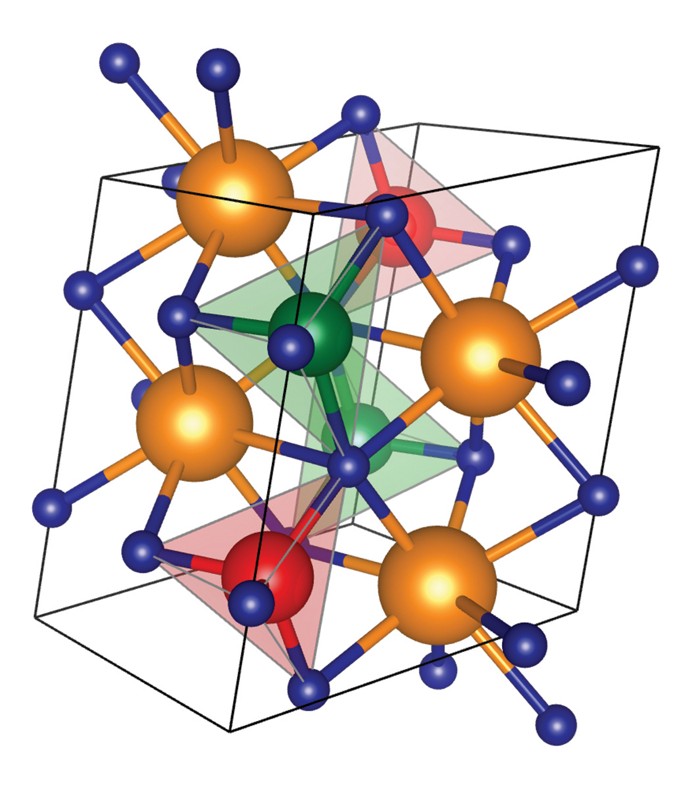
Xingyu’s first paper titled “Water Contributes to Higher Energy Density and Cycling Stability of Prussian Blue Analogue Cathodes for Aqueous Sodium-Ion Batteries” is now published in Chemistry of Materials! In this work, we show that dry Prussian blue analogues (PBAs), one of the most promising cathode materials for aqueous sodium-ion batteries for large-scale energy-storage systems, generally undergo a phase transition from a rhombohedral Na2PR(CN)6 to a tetragonal/cubic PR(CN)6 during Na extraction. However, the presence of water fundamentally alters this phsae behavior, increasing an increase in the average voltage and a reduction in volume change during electrochemical cycling, resulting in both higher energy density and better cycling stability. We also identified four new promising PBA compositions, Na2CoMn(CN)6, Na2NiMn(CN)6, Na2CuMn(CN)6 and Na2ZnMn(CN)6 for further exploration.