Cation Ordering in P2 Na-Ion Cathodes
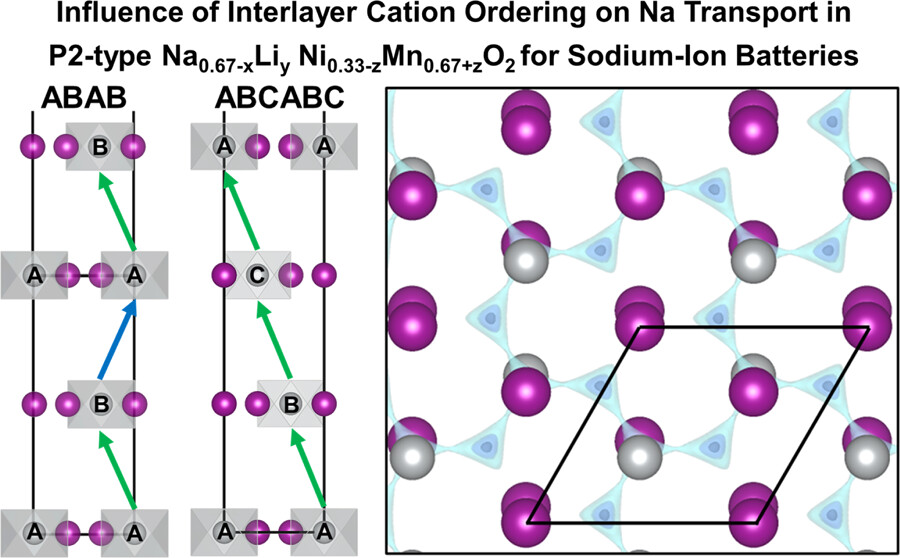
Congratulations to Zishen for his co-authored paper on “Influence of Interlayer Cation Ordering on Na Transport in P2-Type Na0.67–xLiy Ni0.33–zMn0.67+zO2 for Sodium-Ion Batteries” published in JACS together with the group of Prof Claire Xiong at Boise State University! In this work, we studied the P2-type Na2/3Ni1/3Mn2/3O2 (PNNMO) cathode for Na-ion batteries. Zishen’s contribution is showing via DFT calculations that Li doping (Na2/3Li0.05Ni1/3Mn2/3O2, LFN5) promotes ABC-type interplanar Ni/ Mn ordering without disrupting the Na+/vacancy ordering and creates low-energy Li−Mn-coordinated diffusion pathways. These result are in line with those from neutron/X-ray diffraction. Quasielastic neutron scattering reveals that the Na+ diffusivity in LFN5 is enhanced by an order of magnitude over PNNMO, increasing its capacity at a high current. These results suggest that the interlayer ordering can be tuned through the control of composition, which has an equal or greater impact on Na+ diffusion than the Na+/vacancy ordering.
Check out the work here.