

L-edge XANES database
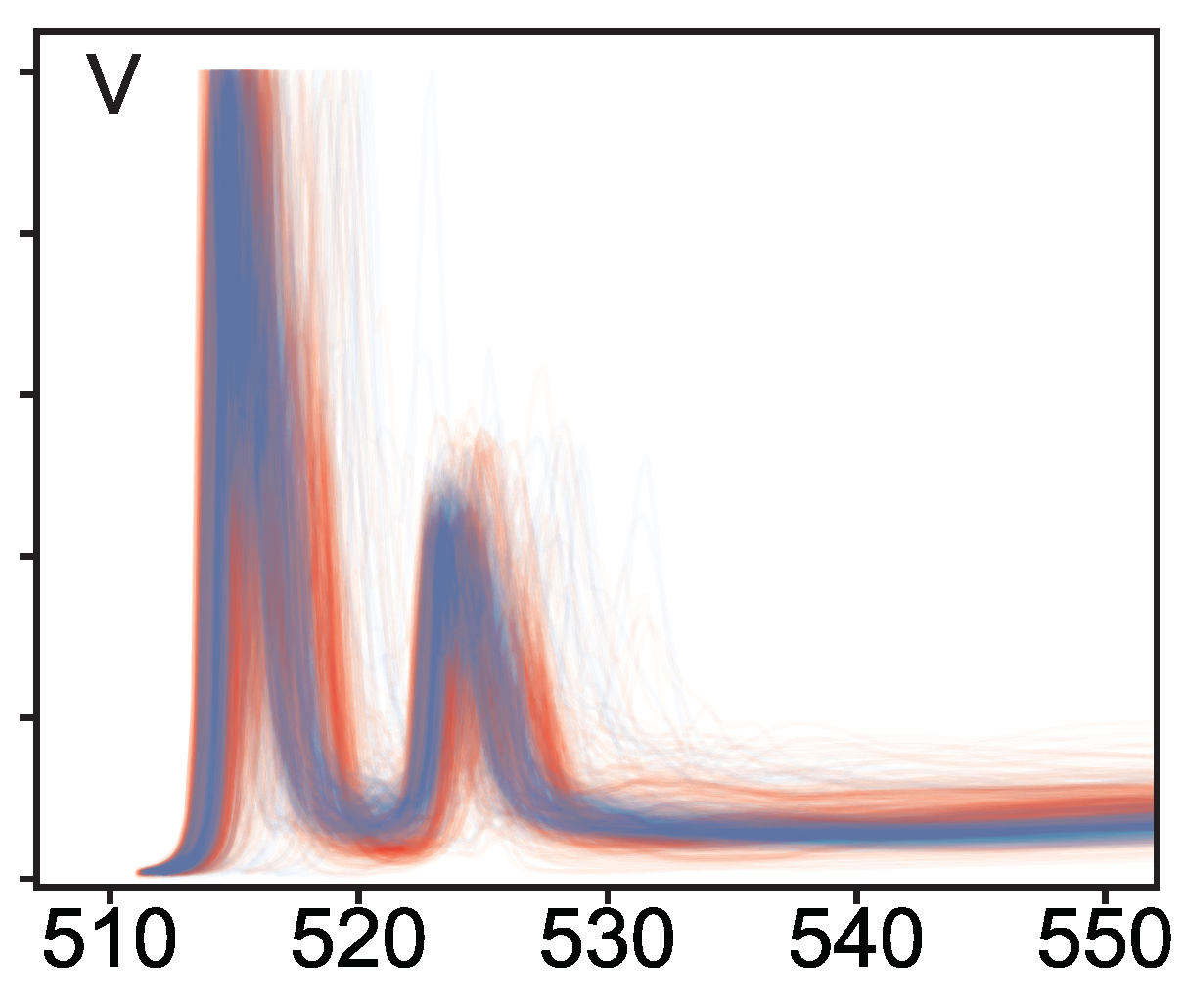
Yiming’s paper on “Database of ab initio L-edge X-ray absorption near edge structure” has just been published in Nature Scientific Data! This work is a collaboration between the Materials Virtual Lab, the Materials Project, Alan Dozier and the groups of Prof John Rehr at the University of Washington and Prof Jordi Cabana at the University of Illinois Chicago. It is a follow-up to our earlier work on a K-edge XANES database, the L-edge XANES database provides instant access to more than 140,000 L-edge spectra for more than 22,000 structures generated using a high-throughput FEFF9 workflow. The L-edge XANES is widely used in the characterization of transition metal compounds. The data is available through the Materials Project XAS app and addresses a critical need for L-edge XANES spectra among the research community. The journal article is available at this link.


{kind=link}
You must be logged in to post a comment.