Unlocking Superionic Sodium Transport and Synthesis with MLIPs
All-solid-state sodium batteries are attractive for grid-scale storage, but the search for a solid electrolyte that combines high ionic conductivity, mechanical compliance, and electrochemical stability has been challenging. In this collaboration with the Meng group published in Joule, we demonstrate that a metastable orthorhombic sodium closo-hydridoborate, Na₃(B₁₂H₁₂)(BH₄) (o-NBH) achieves 4.6 mS cm⁻¹ room-temperature conductivity and enables thick-cathode Na-ASSBs with >3 mAh cm⁻² areal capacity.
Our group’s main contribution to this work is in the application of state-of-the-art machine learning interatomic potentials (MLIPs) in guiding synthesis and probing ion transport. MAVRL member, Zihan Yu, used using high-throughput r2SCAN density-functional theory (DFT) calculations to create a dataset and fine-tune a M3GNet foundation potential for the Na-B-H system. This MLIP allowed the generation of finite-temperature phase diagrams with phonon-derived vibrational entropies. These calculations revealed taht o-NBH sits ~16 meV atom⁻¹ above the 0 K hull but is entropically stabilized above ~650 K, in agreement with experiments.
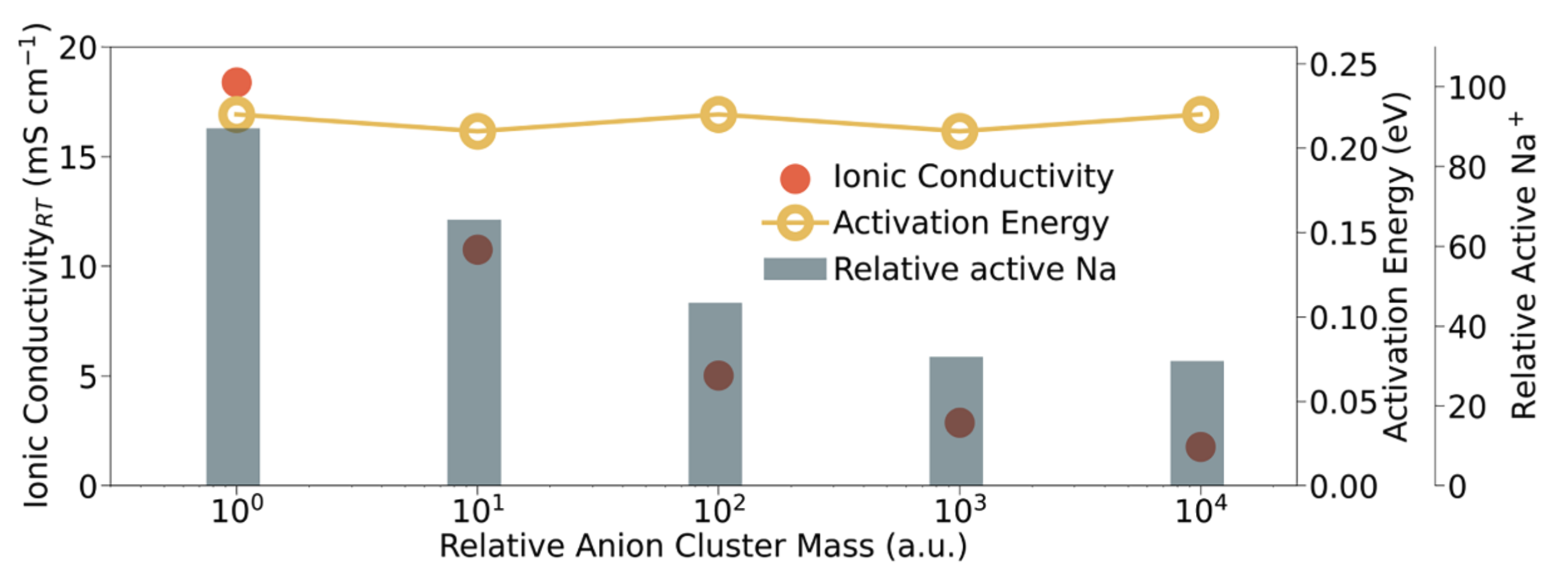
MD simulations with the fine-tuned M3GNet potential shows that o-NBH exhibits 3D Na⁺ diffusion pathways with activation energies of 0.13 eV (short-range) and 0.23 eV (long-range), consistent with NMR relaxation experiments. One major innovation is the use of artificial anion masses to probe the effect of anion rotation on ionic conductivity. Unlike most computational studies that rigidly freeze atoms, our approach provides a more calibrated approach to modify anion rotation dynamics. We find that increased B₁₂H₁₂ or BH₄ cluster masses suppressed Na⁺ conductivity without significantly raising the activation barrier. This suggests that anion rotations boost the population of mobile Na⁺ rather than lowering the hopping barrier.
Check out the work here.